
Your Sample. Our Purpose.
Automation tailored to your workflow – not the other way around!
Don't miss out on any more news!
Sample Preparation
Automation for GC, GC/MS, LC, LC/MS, or standalone
Thermal desorption
Flexible and robust systems for GC/MS
GC-Olfactometry
Detection of aromas, fragrances and off-odors
Personal contact
Direct, individual advice
News from GERSTEL
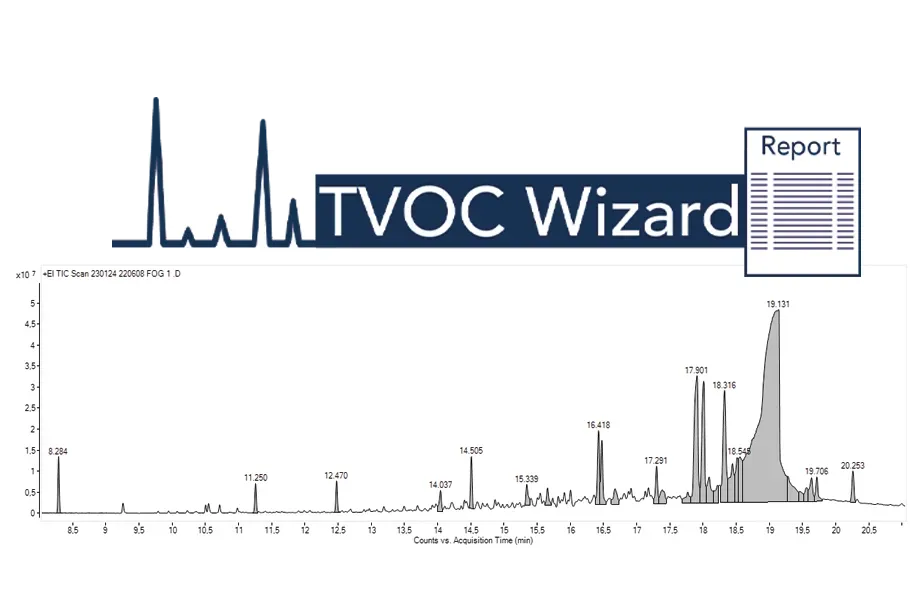
Automated TVOC Wizard Workflow:
Simplifying Data Processing

Utilisation of stir bar sorptive extraction for the analysis of biogenic amines in wines

Sensory Directed Analysis Workshops in Germany, China and Singapore

New AppNote: Sensory-Active Compounds in Cannabis
GERSTEL Applications
Background knowledge in a compact format: While our comprehensive AppNotes provide detailed analysis and technical guidance, the AppBriefs focus on presenting the most important insights and data in a clear, two-page format.

Experience GERSTEL
Our latest video
